Лизосомные болезни: у пациентов появилось надежда
Горовенко Н.Г.
член-корреспондент АМН Украины, профессор, заслуженный деятель науки и техники Украины, заведующая кафедрой медицинской генетики Национальной медицинской академии последипломного образования им. П.Л.Шупика
Лизосомные болезни (ЛБ) - это тяжелые наследственные заболевания, вызванные дефицитом активности специфических ферментов лизосом, которые отвечают за определенный этап деградации сложных комплексов белков, углеводов, липидов.
Вследствие дефектности ферментов внутри лизосом возникает неадекватное накопление субстрата (что и обуславливает другое название этой группы заболеваний - болезни накопления), которое приводит к нарушениям функции клеток различных тканей организма и, как следствие, к появлению клинических симптомов тяжелого прогрессирующего заболевания с поражением многих органов и систем.
Суммарная частота ЛБ, представленных более чем 40 заболеваниями, по последним данным достигает 1:5000 новорожденных. Так, в США ежегодно рождается до 200 000 детей с ЛБ, что показывает важность этой медицинской и социальной проблемы. В Украине
точная частота ЛБ неизвестна вследствие неполной диагностики, что означает установление ложных диагнозов у большинства больных и неправильное лечение с большими финансовыми затратами.
Причиной нарушения активности фермента при ЛБ является генетический дефект. Большинство
генов, мутации в которых приводят к снижению активности лизосомальных ферментов, уже идентифицированы, известна их локализация на определенной хромосоме. Количество разновидностей мутаций в каждом гене достигает сотен, что в ряде случаев затрудняет проведение молекулярной диагностики, хотя число наиболее часто встречаемых в различных популяциях мутаций не превышает 10-20.
Мутации приводят к изменению структуры и, впоследствии, функции кислых гидролаз - ферментов, играющих важнейшую роль в катаболизме макромолекул в лизосомах с оптимумом рН 4-6; нерасщепленный субстрат накапливается в клетках, вследствие
чего резко нарушается их функция. Как правило, активность лизосомальных ферментов у больных не превышает 10-20% от нормы, хотя нет прямой корреляции между уровнем активности ущербного фермента и тяжестью клинических проявлений. В зависимости от характеристики субстрата и поврежденных ферментов строится основная классификация ЛБ, в соответствии с которой выделяют:
- мукополисахаридозы (синдром Гурлера тип I H, синдром Шейе - тип I S, синдром Хантера - тип II, синдром Сан-Филиппо - тип III A, III B , III C, III D, синдром Моркио - тип IY A, IY B, синдром Морото -_Лами - тип YI, синдром Слая - тип YII);
- сфинголипидозы (ганглиозидоз GM1, болезнь Гоше, ганглиозидоз GM2 (Тея-Сакса), метахроматическая лейкодистрофия, болезнь Крабе, болезнь Фарбера, болезнь Шиндлера, болезнь Фабри, Болезнь Нимана-Пика А,В, болезнь Нимана-Пика С);
- муколипидозы, гликопротеинозы и другие ЛБ (болезнь Вольмана, цероидный липофусциноз, муколипидоз І типа (сиалидоз), муколипидоз ІІ типа, муколипидоз ІІІ типа (псевдо Гурлер), маннозидоз).
Диагноз ЛБ можно заподозрить на основании данных клинического обследования: издавна известная характерная внешность с грубыми чертами лица, отображенная в фигурках - гаргоилах, которые украшают собор Парижской богоматери; различные неврологические нарушения; скелетные аномалии; патология глаз и слуха; изменения внутренних органов; различная степень умственной отсталости.
Манифестация симптомов может произойти в разное время - от периода новорожденности до зрелого возраста, в связи с чем различают ранние и поздние
формы заболеваний.
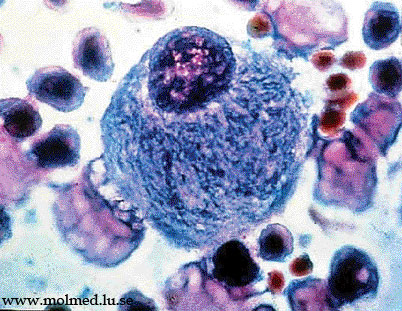
Морфологические исследования направлены на выявление специфических признаков патологического накопления макромолекул в клетках костного мозга, селезенки, нервной ткани, фибробластов кожи. В ряде случаев эти изменения очень типичны
(клетки Гоше, Нимана-Пика).
Основной является биохимическая диагностика, направленная на выявление специфических патологических метаболитов в тканях и жидкостях организма (например, гликозаминогликанов), определение активности ферментов, исследование
меченого радиоизотопами субстрата в фибробластах кожи, а также молекулярно-генетическая диагностика, позволяющая выявить каузальные мутации в соответствующих генах.
Усилия медико-генетического консультирования направлены на диагностику ЛБ, в том
числе и молекулярно-генетическую, установление типа наследования заболевания с последующим расчетом генетического риска рождения больного ребенка в семье и помощи супружеской паре в принятии решения о дальнейшем репродуктивном выборе, учитывая наличие в семье ребенка или родственника с таким
тяжелым заболеванием, как ЛБ.
Обычно повторный риск составляет 25% (при аутосомно-рецессивном типе наследования - наиболее распространенном для большинства ЛБ) или 50% для мальчиков (при Х-сцепленном наследовании - болезни Хантера, Фабри, Данона), хотя обсуждается вероятность наличия заболевание и у лиц женского пола - носителей Х-хромосомы с мутантным геном.
Возможности проведения анализа материала, полученного из биоптата ворсинок хориона, плаценты, или амниотической жидкости, позволяют установить наличие или отсутствие генетического дефекта у конкретного плода, что позволяет супружеским парам высокого риска осознанно принять решение о продолжении или прерывании беременности.
Одной из наиболее известных генетических лизосомальных болезней накопления является болезнь Гоше. Это аутосомно-рецессивное заболевание, в основе которого лежит врожденный дефект незаменимого фермента бета-глюкоцереброзидазы,
вследствие чего метаболический субстрат - глюкоцереброзид (мембранный жир) накапливается в макрофагах (так называемые клетки Гоше), которые массивно инфильтрируют многие органы и системы (печень, селезенку, легкие, костный мозг, кости, центральную и периферическую нервную систему) с нарушением их функций. Хроническое мультисистемное прогрессирующее заболевание.
В клинике преобладают анемия, тромбоцитопения с кровотечениями,
гепатоспленомегалия, поражение скелета с "костными кризисами", отставание в физическом развитии. Выделяют три типа Болезни Гоше: тип 1 - хронический ненейропатический, тип 2 - острый нейропатический, тип 3 - подострый нейропатический. Диагностика основана на определении активности глюкозидазы в лейкоцитах или культурах фибробластов (менее 30% от нормы), а также на определении клеток Гоше в биоптатах костного мозга.
Ранее диагноз ЛБ звучал как приговор, так как никакого специфического лечения не было, ограничивались симптоматической терапией с минимальным объемом реальной помощи пациенту.
В 90-е годы появились первые коммерческие препараты для заместительной ферментотерапии болезни Гоше, которые перевернули всю систему менеджмента ЛБ. У больных появилась надежда. На фоне лечения ферментом имиглюцеразой размеры печени и селезенки уменьшались практически до нормальных, исчезали сильнейшие боли в костях, гемограмма нормализовалась.
Для пациентов и их родителей достигнутое качество жизни стало исполнением несбыточных надежд. На сегодняшний день в мире сотни пациентов с болезнью Гоше на фоне заместительной ферментотерапии ведут обычный образ жизни, им не проводят калечащие операции спленэктомии, являющиеся операцией отчаяния, они учатся в школе, вузе, рожают детей.
Кроме того, разработаны рекомбинантные препараты для лечения мукополисахаридоза типов I и VI, болезни Фабри, которые с успехом применяются во многих странах мира.
В этом веке, наконец, надежда пришла и к пациентам с ЛБ в Украине - благодаря спонсорской помощи первые 15 из них начали получать препарат имиглюцеразы. В текущем году впервые принято решение о государственной закупке препарата для лечения части этих больных.
Литература
Medicus Amicus #3, 2006
|




 Новости
Новости 



