СИНДРОМ АПЕРА: клинические проявления и этиология
Пороки развития черепно-лицевой области занимают 3-е место среди других видов врожденных аномалий. По данным экспертов Всемирной организации здравоохранения (1999), около 7% живорожденных детей имеют врожденные пороки и уродства черепно-лицевой области. Среди врожденных черепно-лицевых деформаций около 30% приходится на краниосиностозы. Из всех синдромальных форм краниосиностозов наиболее часто встречается, по мнению подавляющего большинства специалистов, синдром Апера. В отечественной литературе, к сожалению, часто можно встретить неполную, а иногда и противоречивую информацию о данном синдроме. D. Leibek и C. Olbrich указывают следующие признаки синдрома Апера: дизостозы костей черепа, преждевременный синостоз венечного шва (акроцефалия, высокий шпилеподобный череп), стреловидного шва (скафоцефалия) или других швов; дисморфия лицевого черепа: глазной гипертелоризм, широкий корень носа, щелевидный нос, плоские глазницы, экзофтальм; кожные или костные синдактилии, обычно двусторонние; редко — полидактилия [5]. Ранее считались факультативными признаками лучелоктевые синостозы, синостозы крупных суставов, особенно локтевого, hallux varus, пороки развития позвонков, аплазия акромиоклавикулярных суставов, высокое стояние неба, расщепленный язычок, атрезия заднепроходного отверстия, атрофия зрительного нерва, задержка психического развития, малый рост.
Л. О. Бадалян в своем труде, посвященном описанию клинических проявлений различных синдромов, отмечает, что синдром Апера проявляется изменением формы головы (акроцефалия) и полисиндактилией, большие пальцы ног увеличены в размерах, имеются добавочные большие пальцы, психическое развитие не нарушено [1].
Давая клиническую характеристику синдрома Апера, Х. А. Калмакаров, Н. А. Рабухина, В. М Безруков отмечают, что у синдрома Апера, сочетающего в себе краниофациальный дизостоз с акроцефалией и синдактилией, имеется много общего с дизостозом Крузона [2]. В противоположность дизостозу Крузона, при этом виде дискраний наблюдается раннее синостозирование черепных швов. Этот процесс захватывает все черепные швы, за исключением венечных. Поэтому рост идет преимущественно в высоту, череп приобретает башенную форму и остается узким в переднезаднем и поперечном направлениях. Лоб и затылок широкие и плоские. Как и при дизостозе Крузона, отмечается выраженный экзофтальм из-за уменьшения глубины орбиты и глазной гипертелоризм из-за увеличения размеров решетчатого лабиринта. Верхняя челюсть недоразвита, соотношения зубных рядов нарушены, однако сами зубы развиваются нормально. При синдроме Апера встречается характерная деформация век — они несколько приподняты и образуют складки, поддерживающие глазные яблоки. Наблюдается также птоз верхних век и косоглазие, уплощение носа. Умственное развитие больных с этим синдромом обычно не нарушается, но отмечается очень резкая эмоциональная возбудимость. Характерно сращение нескольких пальцев верхних или нижних конечностей.
С. И. Козлова и соавторы указывают, что синдром Апера характеризуется изменениями черепа — синостоз различной выраженности в основном венечных швов в сочетании со сфеноэтмоидомаксиллярной гипоплазией основания черепа; изменениями лица — плоский лоб, глазной гипертелоризм, антимонголоидный разрез глаз; запавшая переносица, прогнатизм, полное сращение 2–5-го пальцев кистей и стоп [3].
И.Р.Лазовскис описывает синдром Апера как комплекс наследственных аномалий (аутосомно-доминантное наследование): дизостоз черепа — преждевременный синостоз венечного шва (с образованием акроцефалии), ламбдовидного шва (со скафоцефалией), часто преждевременный синостоз всех швов; дисморфия лицевого черепа: глазной гипертелоризм, расширенный корень носа, плоские орбиты, пучеглазие (экзофтальм); кожные или костные синдактилии, обычно двусторонние, реже — полидактилия; изредка наблюдаются синостоз лучевой и локтевой костей и крупных суставов, анкилоз локтевого сустава, аномалии позвоночника, высокое небо, расщепление небного язычка, офтальмоплегия, ослабление зрения; атрезия анального отверстия, умственная отсталость, карликовый рост [4].
Вся эта противоречивая информация, представленная в отечественных источниках, вносит определенную путаницу и усложняет выбор адекватного метода лечения. В основном данные, касающиеся данной темы, отражены в зарубежных источниках.
Клинические проявления синдрома Апера
Основные клинические проявления синдрома акроцефалосиндактилии, описанные французским врачом E. Apert в 1906 г. и названные его именем, сводились к следующему: краниосиностоз, гипоплазия средней зоны лица, симметричная синдактилия кистей и стоп с вовлечением 2–4-го пальцев.
В США распространенность оценивается как 1 на 65 000 (приблизительно 15,5 на 1 000 000) живорожденных. Blank описал собранный материал по 54 пациентам, рожденным в Великобритании [7]. Он разделил пациентов на две клинические категории: «типичная» акроцефалосиндактелия, к которой он применил название «синдром Апера», и другие формы, смешанные в общую группу как «нетипичные» акроцефалосиндактилии. Особенность, отличающая эти типы, — «средний палец», состоящий из нескольких пальцев (обычно 2–4-й), с единственным общим ногтем, наблюдаемый при синдроме Апера и не встречающийся в другой группе. Из этих 54 пациентов 39 имели синдром Апера. Частота синдрома Апера оценивалась им как 1 на 160 000 живорожденных. Cohen и соавторы изучили распространенность случаев рождений с синдромом Апера в Дании, Италии, Испании и частично в Соединенных Штатах [9]. Общее количество дало возможность вывести расчетную частоту рождений с синдромом Апера — приблизительно 15,5 на 1 000 0000 живорождений. Данная цифра превышает примерно вдвое результаты других исследований. Czeizel и соавторы сделали сообщение о частоте рождений больных с синдромом Апера в Венгрии, она составила 9,9 на 1 000 000 живорожденных. Tolarova и соавторы сообщили, что по результатам Калифорнийской программы мониторинга врожденных заболеваний за период с 1983 по 1993 г. было идентифицировано 33 новорожденных с синдромом Апера [26]. Данные были дополнены 22 случаями, описанными в Центре краниофациальных пороков (Сан-Франциско). Частота, определенная на основании этих данных, составила 31 случай на 12,4 млн живорожденных. Больные с синдромом Апера составляют 4,5% всех случаев краниосиностозов. Большинство случаев спорадические и являются следствием новых мутаций, однако в литературных источниках имеется описание семейных случаев с полной пенетрантностью. Weech описал мать и дочь [28], Van den Bosch, по данным Blank [7], наблюдал типичную картину у матери и сына. Rollnick описал больных отца и дочь, что явилось первым примером передачи заболевания по отцовской линии [23]. Данные факты позволяют предположить аутосомно-доминантный тип наследования.
Азиаты имеют самую высокую распространенность синдрома — 22,3 на 1 млн живорождений, испанцы, напротив, самую низкую — 7,6 на 1 млн живорождений [26]. Связь с половой принадлежностью не была выявлена ни одним из исследователей.
Синдром Апера обычно диагностируется в раннем возрасте из-за обнаружения после рождения краниосиностоза и синдактилии. Для синдрома характерно наличие первичных изменений со стороны черепа уже при рождении, однако окончательное формирование патологической формы происходит в течение первых трех лет жизни. У многих пациентов имеется затруднение носового дыхания, из-за сокращения размера носоглотки и хоан, также могут быть затруднения прохождения воздуха через трахею, из-за врожденной аномалии хрящей трахеи, что может привести к ранней смерти. Возможны головная боль и рвота — признаки увеличенного внутричерепного давления, особенно в случаях, когда в процесс вовлечено несколько швов. Генеалогический анамнез представляется не столь важным, поскольку большинство случаев рождений детей с данным синдромом являются спорадическими.
Фенотипические признаки синдрома Апера
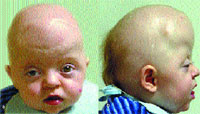 |
| Рисунок 1. Внешний вид больного в возрасте 1,5 года с синдромом Апера: анфас (а) и профиль (б) |
Черепно-лицевая область. Наиболее часто встречается коронарный краниосиностоз, приводящий к акроцефалии, брахицефалии, туррибрахицефалии. Синостозированию подвергаются также сагиттальные, ламбдовидные, лобно-основные швы. Редкая аномалия черепа в виде трилистника найдена приблизительно у 4% младенцев. Основание черепа уменьшено в размерах и часто асимметрично, передняя черепная ямка очень короткая. Передний и задний роднички увеличены в размерах и не заращены. Средняя линия свода черепа может иметь зияющий дефект, простирающийся от области глабеллы через область метопического шва до переднего родничка, через область сагиттального шва до заднего родничка. Отмечаются: глазной гипертелоризм, экзорбитизм, мелкие орбиты, нависающие надбровные дуги. Со стороны глаз наблюдаются: экзофтальм, «прерывистые брови», пальпебральные трещины, косоглазие, амблиопия, атрофия зрительного нерва, и (редко) вывих глазного яблока, снижение пигмента, врожденная глаукома, обратимая потеря зрения. Переносица часто запавшая. Нос короткий с уплощенной спинкой и с широким кончиком со стенозом или атрезией хоан, носогубные складки глубокие, возможна девиация носовой перегородки. Имеется гипоплазия средней зоны лица — верхняя челюсть гипоплазирована, скуловые дуги короткие, скуловые кости мелкие. В связи с этим отмечается относительный нижнечелюстной прогнатизм. Рот в состоянии покоя имеет трапециевидную форму. Высокое аркообразное небо, расщелина мягкого неба и язычка наблюдается в 30% случаев. Твердое небо короче, чем в норме, мягкое небо — длиннее и толще, верхнечелюстная зубная дуга имеет V-образную форму. Могут быть выступающие из ряда верхние зубы, имеющие форму совка резцы, сверхкомплектные зубы и выступающие альвеолярные гребни. Пациенты имеют низко посаженные уши и высокую вероятность снижения слуха в дальнейшем (рис. 1, 2).
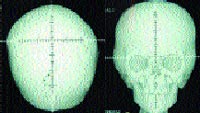 |
| Рисунок 2. Рентгенограмма черепа больного с синдромом Апера в прямой проекции (формат 3 D) |
Конечности и скелет. Одним из основных проявлений синдрома является синдактилия кистей и стоп с вовлечением 2, 3 и 4-го пальцев. Реже в процесс вовлекаются 1-й и 5-й пальцы (рис. 3). Проксимальные фаланги больших пальцев кистей и стоп укорочены, дистальные имеют трапециевидную форму. При изучении синдрома Апера Wilkie и соавторы [30] внесли изменения в классификацию синдактилий Upton (1991). При синдроме Апера центральные три пальца всегда подвергнуты синдактилии. Тип 1 — большой палец и часть 5-го пальца отделены от сросшихся пальцев; при типе 2 — только большой палец отделен от «среднего пальца»; при типе 3 — все пальцы сросшиеся. Точно так же синдактилия пальцев стопы может вовлекать три боковых пальца (тип 1), или 2–5-й пальцы с отдельным большим пальцем ноги (тип 2), или может быть непрерывной (тип 3). Cohen и Kreiborg изучили 44 пары рук и 37 пар ног пациентов с синдромом Апера, используя клинический, радиографический методы и дерматоглифику [10], а также изучили гистологические препараты верхних конечностей мертворожденного плода со сроком 31 нед. Они предположили, что различие между акроцефалосиндактилией и акроцефалополисиндактилией является ложным и что от использования этих терминов следует отказаться. Исследователи также указали на то, что при синдроме Апера патология верхних конечностей всегда более выражена, чем нижних. Сращение костей запястья с дистальными фалангами не имеет своего аналога на стопе. Возможны и другие патологические изменения конечностей: радиальное отклонение коротких и широких больших пальцев, из-за измененной проксимальной фаланги — брахидактилия; ограничение подвижности в плечевом суставе, ограниченная подвижность локтевого сустава с затруднением пронации и супинации, ограничение подвижности в коленном суставе, аплазия или анкилоз плечевого, локтевого и тазобедренного сустава. Одной из сравнительно часто встречаемых аномалий скелета при синдроме Апера является врожденное сращение позвонков. Kleiborg и соавторы обнаружили, что сращение позвонков в шейном отделе наблюдалось у 68% пациентов с синдромом Апера: единичные сращения у 37% и множественные сращения у 31% [13]. Наиболее характерно было сращение C5–C6. Напротив, сращение в шейном отделе происходит только у 25% пациентов с синдромом Крузона и наиболее часто изменены C2–C3. Kleiborg и соавторы сделали заключение, что сращение C5–C6 более характерно для синдрома Апера, а C2–C3 для синдрома Крузона, что помогает дифференцировать эти два заболевания [13]. Рентгенографическое исследование шейного отдела позвоночника является обязательным перед анестезиологическим пособием для этих пациентов. Schauerte и St-Aubin показали, что прогрессивный синостоз отмечается не только в черепных швах, но и в костях ног, рук, запястьях, шейном отделе позвоночника и предложили термин «прогрессирующий синостоз с синдактилией» как наиболее адекватно отражающий клиническую картину [24].
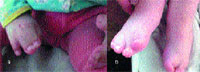 |
| Рисунок 3. Вид верхних (а) и нижних (б) конечностей больного С. с синдромом Апера |
Кожа. По некоторым данным, для синдрома Апера характерны элементы глазо-кожного альбинизма (светлые волосы и бледная окраска кожных покровов). Cohen и Kreiborg описали кожные проявления в 136 случаях синдрома [10]. Они обнаружили гипергидроз у всех пациентов. Также они описали акнеформные элементы, которые были особенно распространены на лице, груди, спине, руках. Помимо этого возможны проявления гипопигментации и гиперкератоза ладоней, западения кожи над крупными суставами конечностей. У некоторых пациентов имеется избыточная кожа складок лба.
Центральная нервная система (ЦНС). С синдромом связаны различные степени умственного дефицита, однако есть сообщения и о больных с нормальным интеллектом. Повреждения ЦНС в большинстве случаев могут быть причиной умственной отсталости. Возможно, проведение краниоэктомии на ранних этапах способствует нормальному умственному развитию. Patton и соавторы [20] проводили долгосрочное исследование 29 пациентов, из которых 14 имели нормальный или пограничный показатель интеллекта, у 9 отмечалась незначительнуая умственная отсталость (коэффициет интеллекта (IQ) 50–70), 4 были умеренно отсталыми (IQ 35–49) и 2 были выраженно отсталыми (IQ меньше 35). Ранняя краниоэктомия, казалось, не улучшала интеллектуальный статус. Шесть из 7 пациентов, окончивших школу, были приняты на работу или проходили дальнейшее обучение. Вопреки этим заключениям, Park и Powers, Cohen и Kreiborg утверждают, что многие из пациентов умственно отсталые [11, 19]. Они собрали информацию по 30 пациентам с патологией мозолистого тела, или структур лимба, или того и другого. Также у данных больных имелись и другие разнообразные нарушения. Авторы предположили, что эти аномалии могут быть причиной умственной отсталости. Прогрессирующая гидроцефалия встречалась редко, и часто ее не удавалось дифференцировать с непрогрессирующей вентрикуломегалией. Cinalli и соавторы обнаружили, что только 4 из 65 пациентов с синдромом Апера были шунтированы в связи с прогрессирующей гидроцефалией [8]. Renier и соавторы нашли уровень интеллекта 70 и больше у 50% детей из тех, кто имел декомпрессию черепа до 1 года, против 7,1% из тех, кто перенес оперативное лечение в позднем возрасте [22]. Патология corpus callosum (мозолистое тело) и размер желудочков мозга не коррелировались с заключительным показателем интеллекта, в отличие от патологии septum pellucidum (прозрачная перегородка). Качество окружающей среды и семейное окружение также определяют интеллектуальное развитие. Только 12,5% детей с данным синдромом имеют нормальные показатели интеллекта, по сравнению с 39,3% детей с нормальным внутрисемейным фоном.
Внутренние органы и системы. Для синдрома Апера характерны незначительные изменения со стороны внутренних органов. Патология со стороны сердечно-сосудистой системы (дефект межжелудочковой перегородки, несращенный Баталлов проток, стеноз легочной артерии, коарктация аорты, декстракардия, тетрада Фалло, эндрокардиальный фиброэластоз) отмечается у 10–20% больных. Аномалии мочеполовой системы (поликистоз почек, добавочные почечные лоханки, гидронефроз, стеноз шейки мочевого пузыря, двурогая матка, атрезия влагалища, увеличенные большие половые губы, клиторомегалия, крипторхизм) выявлены у 9,6%. Аномалии пищеварительной системы (пилоростеноз, атрезия пищевода, эктопия заднего прохода, частичная атрезия или недоразвитие желчного пузыря) обнаружены у 1,5%. Pelz и соавторы описали 18-месячную девочку, которая имела дистальный эзофагальный синдром в дополнение к типичным проявлениям синдрома Апера. Также в литературе упоминаются патологические изменения дыхательной системы — аномальные хрящи трахеи, трахеопищеводный свищ, легочная аплазия, отсутствие средней доли легкого, отсутствующие междолевые борозды [6, 11, 15].
Этиология синдрома Апера
За редкими исключениями синдром Апера вызывается одной из двух миссенс-мутаций гена FGFR2, вовлекающей две смежные аминокислоты: S252W и P253R, у 63% и 37% пациентов соответственно, по данным Wilkie и соавторов [29]. Park и соавторы исследовали корреляции фенотип/генотип у 36 больных с синдромом Апера [19]. Почти у всех, за исключением одного пациента, были найдены мутации S252W или P253R в гене FGFR2; частота составила 71 и 26% соответственно. Факт, что один пациент не имел мутации в этой области, дает основание предполагать наличие генетической гетерогенности синдрома Апера. Изучение 29 различных клинических проявлений продемонстрировало статистически несущественные различия между двумя подгруппами пациентов, имевших две основные мутации. Moloney и соавторы предоставили информацию относительно спектра мутаций и наследственного характера мутаций при синдроме Апера [16]. Их анализ 118 пациентов показал, что мутационный спектр при синдроме Апера узок. Мутация S252W наблюдалась у 74, а P253R — у 44 пациентов. Slaney и соавторы обнаружили отличия между клиническими проявлениями синдактилии и небной расщелины при двух основных мутациях гена FGFR2 при синдроме Апера [25]. Среди 70 пациентов с синдромом Апера 45 имели мутацию S252W и 25 — мутацию P253R. Синдактилия кистей и стоп была более серьезно выражена у пациентов с мутацией P253R. Напротив, расщелины неба оказались более характерны для пациентов с мутацией S252W. Различий в проявлении других патологий, связанных с синдромом Апера, найдено не было. Lajeunie и соавторы проводили скрининговое исследование 36 пациентов с синдромом Апера в целях обнаружения мутаций в гене FGFR2 [14]. Мутации были обнаружены во всех случаях. У 23 пациентов (64%) была обнаружена мутация ser252trp. У 12 пациентов (33%) была выявлена мутация pro253arg. Oldridge и соавторы проанализировали истории болезни 260 неродственных пациентов с синдромом Апера и нашли, что 258 имели миссенс-мутацию в экзоне 7 гена FGFR2, которая повреждала белок в линкерном районе между вторыми и третьими иммуноглобулиноподобными доменами [17]. Следовательно, генетическая причина возникновения синдрома Апера достаточно точно определена. Авторы установили, что 2 пациента имели вставки Alu-элемента в экзоне 9 или около него. Изучение фибробластов показало эктопическую экспрессию KGFR области FGFR2, которая была связана с выраженностью патологий конечностей. Эта корреляция оказалась первым генетическим свидетельством того, что аномальная экспрессия KGFR является причиной синдактилии при синдроме Апера. Основные миссенс-мутации в экзоне 7 (ser252trp и ser252phe) были выявлены у 258 и 172 пациентов соответственно. Von Gernet и соавторы проводили исследования относительно постхирургических проявлений в черепно-лицевой области у больных с различной степенью синдактилии [27]. У 21 пациента с синдромом Апера, из тех, кто подвергся хирургическому лечению краниофациальной области, лучшая клиническая картина была у больных с мутацией P253R, хотя они имели более серьезную форму синдактилии. Мутация P253R была определена у 6, а S252W — у 15 пациентов.
Диагностика и лечение
Удалось доказать, что больше чем 98% случаев вызваны определенными миссенс-мутациями, вовлекающими смежные аминокислоты (Ser252Trp, Ser252Phe или Pro253Arg) в экзоне 7 гена FGFR2, в связи с чем появилась возможность молекулярно-генетической диагностики синдрома Апера. Пока же этот метод не получил широкого распространения, основным способом диагностики является проведение компьютерной томографии (КТ) черепа. При помощи КТ выявляются такие характерные патологические изменения костей черепа, как коронарный синостоз, гипоплазия верхней челюсти, мелкие орбиты, изменения основания черепа и т. д. Наиболее наглядными являются данные, полученные при проведении КТ в формате ЗD. Магнитно-резонансная томография (МРТ) помогает оценить изменения мягких тканей черепа, связанные с костной патологией. Также для уточнения клинических проявлений синдрома Апера проводятся рентгенологические исследования костей верхних и нижних конечностей, целью которых является обнаружение различных форм костных синдактилий и изменений костей стоп и кистей. Помимо вышеперечисленных исследований, в диагностике степени выраженности фенотипических проявлений синдрома Апера и для прогноза развития заболевания важны данные психометрической оценки, исследования слуха, состояния дыхательных путей, а кроме того, заключения таких специалистов, как педиатр, клинический генетик, нейрохирург, ортодонт, отоларинголог, офтальмолог, невролог, психолог, логопед.
Хирургическое лечение включает в себя раннюю краниоэктомию коронарного шва и фронто-орбитальную репозицию для уменьшения проявлений дисморфизма и патологических изменений формы черепа. Операции по поводу синдрома Апера часто состоят из нескольких этапов, последний проводится в подростковом возрасте. Первый этап часто выполняется уже в 3 мес.
В последнее время стала широко использоваться новая техника краниофацильной дистракции с постепенным вытяжением кости. Этот метод приводит к хорошим косметическим результатам и снимает необходимость проведения костной пластики у пациентов в возрасте 6–11 лет. Помимо хирургического лечения патологии костей черепа, пациентам с синдактилией кистей и стоп проводится хирургическое лечение пальцев конечностей. Для формирования физиологического прикуса детям с синдромом Апера назначается ортодонтическое лечение.
Успехи в молекулярной генетике и неуклонное развитие клеточной биологии делают возможным понимание механизмов пороков развития у людей и их пренатальной диагностики. Определение фенотипа и генотипа и их корреляция очень важны для врача. Знание всех клинических проявлений того или иного синдрома позволяет хирургу выбрать правильную тактику ведения больных в пред- и послеоперационном периоде; помогает определить круг специалистов и исследований, необходимых для обследования пациентов. Практика показывает, что проблему лечения больных с синдромальными краниосиностозами нельзя решить при помощи изолированной работы краниофациальных хирургов. Как видно на примере синдрома Апера, синдромальные краниосиностозы сопровождаются не только деформацией костей черепа, но и патологическими изменениями как всего комплекса органов и тканей головы, так и костей скелета и внутренних органов. Для адекватного лечения больных с синдромальными формами краниосиностозов необходимо привлечение нейрохирургов, детских хирургов, педиатров, психологов, неврологов, окулистов, рентгенологов, отоларингологов, логопедов и генетиков. Наилучшие результаты достигаются при объединении усилий врачей всех перечисленных специальностей.
Литература
- Наследственные болезни: справочник. Ташкент: Медицина, 1980. С. 209.
- Калмакаров Х. А., Рабухина Н. А., Безруков В. М. Деформации лицевого черепа. М.: Медицина, 1981. С. 72–96.
- Козлова С. И., Семанова Е., Демикова Н. С., Блинникова О. Е. Наследственные синдромы и медико-генетическое консультирование. М.: Медицина, 1987. С. 14–16.
- Лазовскис И. Р. 2668 клинических симптомов и синдромов. М., 1995. С. 80.
- Leibek D., Oldbrich C. Клинические синдромы: пер. с англ. Л. С. Рабен. М.: Медицина, 1974. С. 23.
- Apert M. E. De l’acrocephalosyndactylie//Bull. Mem. Soc. Med. Hop. 1906; 23: 1310–1330.
- Blank C. E. Apert’s syndrome (a type of acrocephalosyndactyly) — observations on a British series of thirty-nine cases//Ann. Hum. Genet. 1960; 24: 151–164.
- Cinalli G., Renier D., Sebag G., Sainte-Rose C., Arnaund E., Pierre-Kahn A. Chronic tonsillar herniation in Crouzon’s and Apert’s syndromes: the role of premature synostosis of the lambdoid suture// J. Neurosurg. 1995; 83 (4): 575–582.
- Cohen M. M. Jr., Kreiborg S. Lammer E. J., Cordero J. F. et al. Birth prevalence study of the Apert syndrome//Am. J. Med. Genet. 1992; 1: 42 (5): 655–659.
- Cohen M. M., Kreiborg S. Hands and feet in the Apert syndrome//Am. J. Med. Genet. 1995; 22: 57 (1): 82–96.
- Cohen M. M., Kreiborg S. The central nervous system in the Apert syndrome//Am. J. Med. Genet. 1990; 35 (1): 36–45.
- Kreiborg S., Cohen M. Is craniofacial morphology in Apert and Crouzon syndromes the same?//Acta. Odontol. Scand. 1998; 56 (6): 339–341.
- Kreiborg S., Barr M., Cohen M. M. Cervical spine in the Apert syndrome//Am. J. Med. Genet. 1992; 43 (4): 704–708.
- Lajeunie E., Cameron R., El Ghouzzi V., de Parseval N., Journeau P., Gonzales M., Delezoide A. L., Bonaventure J., Le Merrer M., Renier D. Clinical variability in patients with Apert’s syndrome// J. Neurosurg. 1999; 90 (3): 443–447.
- Marsh J., Galic M., Vannier M. Surgical correction of the craniofacial dysmorphology of Apert syndrome//Clin. Plast. Surg. 1991; 18 (2): 251–258.
- Moloney D. Hunterian Lecture. What can we learn about mechanisms of mutation from a study of craniosynostosis?//Ann. R. Coll. Surg. Engl. 2001; 83 (1): 1–9.
- Oldridge M., Zackai E. H., McDonald-McGinn D. M., Iseki S. et al. De novo alu-element insertions in FGFR2 identify a distinct pathological basis for Apert syndrome//Am. J. Hum. Genet. 1999; 64 (2): 446–461.
- Park W. J., Meyers G. A., Li X. et al. Novel FGFR2 mutations in Crouzon and Jackson-Weiss syndromes show allelic heterogeneity and phenotypic variability//Hum. Mol. Genet. 1995; 4 (7): 1229–1233.
- Park E. A., Powers G. F. Acrocephaly and scaphocephaly with symmetrically distributed malformations of the extremities//Am. J. Dis. Child. 1920; 20: 235–315.
- Patton M. A., Goodship J., Hayward R., Lansdown R. Intellectual development in Apert’s syndrome: a long term follow up of 29 patients//J. Med. Genet. 1988; 25(3): 164–167.
- Pelz L., Unger K., Radke M. Esophageal stenosis in acrocephalosyndactyly type I//Am. J. Med. Genet. 1994; 53 (1): 91.
- Renier D., Arnaud E., Cinalli G., Sebag G. et al. Prognosis for mental function in Apert’s syndrome//J. Neurosurg. 1996; 85(1): 66–72.
- Rollnick B. Male transmission of Apert syndrome//Clin. Genet. 1988; 33 (2): 87–90.
- Schauerte E. W., St-Aubin P. M. Progressive synosteosis in Apert’s syndrome (acrocephalosyndactyly), with a description of roentgenographic changes in the feet//Am. J. Roentgenol. Radium. Ther. Nid. Med. 1996; 97 (1): 67–73.
- Slaney S. F., Oldridge M., Hurst J. A., Moriss-Kay G. M. et al. Differential effects of FGFR2 mutations on syndactyly and cleft palate in Apert syndrome//Am. J. Hum. Genet. 1996; 58 (5): 923–932.
- Tolarova M. M., Harris J. A., Ordway D. E., Vargervik K. Birth prevalence, mutation rate, sex ratio, parents’ age, and ethnicity in Apert syndrome//Am. J. Med. Genet. 1997; 72 (4): 394–398.
- Von Gernet S., Golla A., Ehrenfels Y., Schuffenhauer S., Fairley J. D. Genotype-phenotype analysis in Apert syndrome suggests opposite effects of the two recurrent mutations on syndactyly and outcome of craniofacial surgery//Clin. Genet. 2000; 57(2): 137–139.
- Weech A. A. Combined acrocephaly and syndactylism occurring in mother and daughter: a case report//Bull. Johns. Hopkins. Hosp. 1927; 40: 73–76.
- Wilkie A. O. Fibroblast growth factor receptor mutations and craniosynostosis: three receptors, five syndromes//Indian. J. Pediatr. 1996; 63 (3): 351–356.
- Wilkie A. O. M., Slaney S. F., Oldridge M., Poole M. D., Ashworth G. J., Hockley A. D., Hayward R. D., David D. J., Pulleyn L. J., Rutland P., Malcolm S., Winter R. M., Reardon W. Apert syndrome results from localized mutations of FGFR2 and is allelic with Crouzon syndrome//Nature. Genet. 1995; 9 (2): 165–172.
Д. Е. Колтунов, кандидат медицинских наук НПЦ медицинской помощи детям с пороками развития черепно-лицевой области и врожденными заболеваниями нервной системы, Москва
lvrach.ru
www.medicusamicus.com
|




 Новости
Новости 








